![]() Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Time Stamps:
- Â Should patients be screened …

![]() Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Time Stamps:
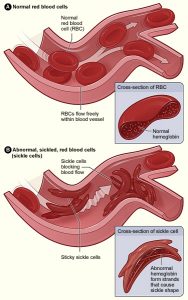 By Leonard Naymagon, MD
By Leonard Naymagon, MD
Peer Reviewed
Vaso-occlusive crisis (VOC), or pain crisis, is the most common clinical manifestation of sickle cell disease (SCD) and is responsible for the majority of emergency department (ED) visits and inpatient …
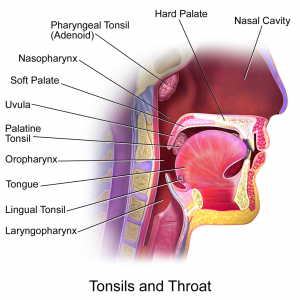 By Tyler Litton, MD
By Tyler Litton, MD
Peer Reviewed
Oropharyngeal squamous cell carcinoma (OPSCC) is relatively rare but incidence has increased in the US over the past 40 years. [1] Tonsillar cancer is the …
 Joshua Horton
Joshua Horton
Peer Reviewed
We are not winning the war against cancer, if war is even an appropriate metaphor. When Richard Nixon signed the National Cancer Act into effect in 1971, many …
![]() By Susanna Jeurling
By Susanna Jeurling
Peer Reviewed
The U.S. Preventive Services Task Force (USPSTF) recently finalized its position regarding annual low-dose computed tomography (LDCT) scanning for early detection of lung cancer. The grade B …
 Please enjoy this post from the archives dated September 8, 2011
Please enjoy this post from the archives dated September 8, 2011
By David Altszuler, Class of 2012
Faculty Peer Reviewed
An empiric association between occult malignancy and thrombophlebitis has been recognized since Trousseau first reported the syndrome in 1865.[1] The …
 By Gabriel Schneider, MD
By Gabriel Schneider, MD
Peer Reviewed
The new oral anticoagulants (NOACs) are an appealing alternative to the burdensome vitamin K antagonists such as warfarin. These novel agents include direct thrombin inhibitors such …
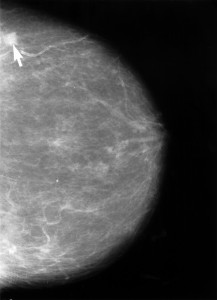 By Miguel A. Saldivar, MD
By Miguel A. Saldivar, MD
Peer Reviewed
Mammograms are far from strangers to the paparazzi. It was only recently that a television news reporter from a prominent broadcasting company reluctantly …