 By Daniela Rebollo Salazar
By Daniela Rebollo Salazar
Peer Reviewed
In the past ten years, the number of bacterial pathogens resistant to multiple antibiotics has dramatically increased. The emergence of resistant microorganisms is a …

By Daniela Rebollo Salazar
Peer Reviewed
In the past ten years, the number of bacterial pathogens resistant to multiple antibiotics has dramatically increased. The emergence of resistant microorganisms is a …
 By Helen Ma, MD
By Helen Ma, MD
Peer Reviewed
Our new Spotlight series uses case vignettes to explore diagnosis, pathophysiology, and management of a wide variety of diseases seen in the outpatient and inpatient settings. Articles in …
 By Martin Fried, MD
By Martin Fried, MD
Peer reviewed
Learning Objectives
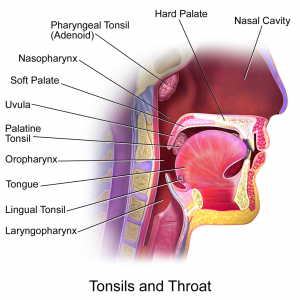 By Tyler Litton, MD
By Tyler Litton, MD
Peer Reviewed
Oropharyngeal squamous cell carcinoma (OPSCC) is relatively rare but incidence has increased in the US over the past 40 years. [1] Tonsillar cancer is the …
 By Gabriel Campion
By Gabriel Campion
Peer Reviewed
For over a century, neckties have been a staple accessory in the wardrobe of the American professional man. Although white-collar dress codes have trended toward a …
 By Ofole Mgbako, MD
By Ofole Mgbako, MD
Peer Reviewed
In July 2010, the much-anticipated “National HIV/AIDS Strategy for the United States†was released to the public. In its introduction, the president declared, “Our Nation is at …
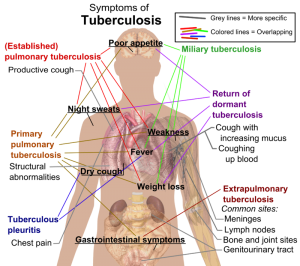 By: Miguel A. Saldivar, MD
By: Miguel A. Saldivar, MD
Peer ReviewedÂ
“Indeterminate.” Many clinicians have expressed frustration when reading this word on a Quantiferon-TB Gold test result. The obligate follow-up question is: what is the next best …
 By M tanner
By M tanner
Many  bacteria live in and on me—I’ve always known that. But when I learned that bacteria make up 90% of the cells in my body, it made me feel so sucio, so unclean.
I went through my …