
 By Kerrilynn Carney, MD
By Kerrilynn Carney, MD
Peer Reviewed
Coronary heart disease remains the leading cause of death globally despite the use of statin therapy. Although major statin studies suggest an average 31% reduction in relative risk of coronary events, a residual risk of 69% remains to be addressed. (1) The search for a medical therapy to ameliorate residual risk has become the holy grail of cardiologists and pharmaceutical companies alike. While high-density lipoprotein cholesterol levels (HDL) independently predict cardiovascular disease risk, interventions to raise circulating HDL levels have thus far been unsuccessful in reducing the risk of coronary heart disease. (2)
When controlling for LDL cholesterol, the odds of coronary heart disease fall by about 40% per 7.5 milligram per deciliter rise in HDL cholesterol (3). While this translates to a fall in major adverse cardiovascular events by about 1.1% for each 1 milligram per deciliter rise in HDL cholesterol when LDL cholesterol is 70 mg/dL, attempts at raising HDL cholesterol with pharmacotherapy in the post-statin era have not produced a similar risk reduction. (4)
HDL is a complex molecule with anti-oxidative, anti-inflammatory, anti-thrombotic, antiplatelet, and vasodilatory properties, which help protect LDL from oxidation. Each HDL particle carries between two and five molecules of apolipoprotein A1 (ApoA-I), over 80 proteins, and hundreds of lipid moieties, with many molecules containing antioxidant and antithrombotic properties (5-7).
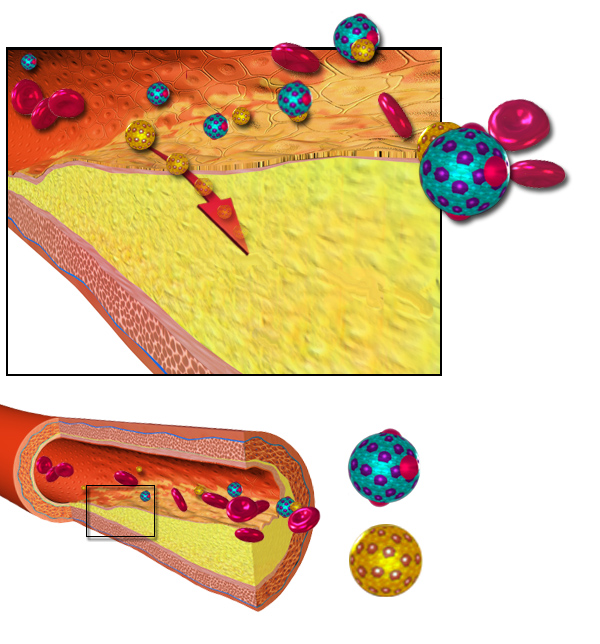
There are multiple mechanisms by which HDL mitigates the process of atherogenesis, but the efflux capacity, or ability to relocate cholesterol out of macrophages, is believed to be central in reducing major adverse cardiovascular events. In macrophages in atherosclerotic lesions, enlargement of intracellular lipid droplets occurs when cholesterol delivery and storage is greater than cholesterol removal, promoting transformation into foam cells. HDL participates in cholesterol efflux via reverse cholesterol transport, relocating cholesterol from peripheral cell membranes via scavenger receptor class B type 1 to hepatocytes centrally. (8) ATP-binding cassette transporter A-1, present in macrophages, the liver, and the intestine, actively transports cholesterol to lipid-poor apolipoprotein A-I particles carried on HDL. In addition, HDL has a major effect on endothelial function, promotes endothelial repair and progenitor cell health, and also supports production of nitric oxide. (7)
There are several well-known examples in which HDL cholesterol levels have been raised without conferring cardio-protection. In preclinical studies, overexpression of hepatic ATP-binding cassette transporter A-1 in an animal model result in higher HDL cholesterol levels, but impaired HDL function and accelerated atherosclerosis. (9) Cholesteryl ester transfer protein (CETP) deficiency from mutations in the CETP gene also increases HDL cholesterol, but evidence varies about their protection against atherosclerosis. Conversely, patients with a specific genetic ezyme deficiency (lecithin cholesterol acyltransferase) or mutations within the ApoA-I gene resulting in hypo-function of apolipoprotein A-I secretion have marked HDL cholesterol deficiency but do not necessarily develop atherosclerosis at a higher rate than normal patients. (10)
HDL augmentation with pharmacotherapy has been wrought with both equivocal and negative studies. Specifically, raising HDL with estrogen (11, 12), fibrates (13, 14), niacin, or CETP inhibitors (15) has not improved outcomes, and in some cases increased adverse events. After the equivocal AIM-HIGH trial of niacin, the HPS2-THRIVE trial showed that among participants with atherosclerotic vascular disease, the addition of extended-release niacin-laropiprant did not significantly reduce the risk of MCVE and actually increased the risk of serious adverse events when compared to statin-based LDL cholesterol-lowering therapy alone (16). CETP inhibitors as a class have variable efficacy in raising HDL levels and conferring cardio-protection, and additionally have shown some evidence for increased harm. (15, 17-19)
The fact remains that when LDL cholesterol is aggressively lowered, low HDL cholesterol levels are inversely related to major coronary vascular events. Raising HDL cholesterol has been a major target in the quest to reduce residual cardiovascular risk in the post-statin era; however raising HDL with pharmacotherapy has not meaningfully effected clinical endpoints and in some cases produced harm. There is a growing impression that HDL functionality, rather than abundance, is clinically important. (7) HDL efflux capacity has been inversely associated with both prevalent and incident cardiovascular disease risk, independent of established cardiovascular risk factors, even after adjusting for HDL cholesterol or apoA-I concentrations, and is an area of active research in the field of lipidology. (20-23)
For now, we should continue using validated risk models to prescribe statin therapy to those who need it and encourage our patients to attempt to raise their HDL levels through diet and exercise.
Dr. Kerrilynn Carney is a 2nd year resident at NYU Langone Medical Center
Peer reviewed by Alana Choy-Shan, MD, Cardiology, NYU Langone Medical Center
Image courtesy of Wikimedia Commons
References
- Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670-1681. http://www.ncbi.nlm.nih.gov/pubmed/21067804
- Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, et al. Major lipids, apolipoproteins, and risk of vascular disease. Jama 2009;302:1993-2000. http://www.ncbi.nlm.nih.gov/pubmed/19903920
- Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, Grundy SM, Kastelein JJ, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 2007;357:1301-1310. http://www.ncbi.nlm.nih.gov/pubmed/17898099
- Carey VJ, Bishop L, Laranjo N, Harshfield BJ, Kwiat C, Sacks FM. Contribution of high plasma triglycerides and low high-density lipoprotein cholesterol to residual risk of coronary heart disease after establishment of low-density lipoprotein cholesterol control. Am J Cardiol 2010;106:757-763. http://www.ncbi.nlm.nih.gov/pubmed/20816113
- Heinecke JW. The protein cargo of HDL: implications for vascular wall biology and therapeutics. J Clin Lipidol 2010;4:371-375. http://www.ncbi.nlm.nih.gov/pubmed/20975842
- Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol 2011;8:222-232.
- Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag 2013;9:617-670.
- Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res 1999;40:781-796.
- Feng Y, Lievens J, Jacobs F, Hoekstra M, Van Craeyveld E, Gordts SC, Snoeys J, et al. Hepatocyte-specific ABCA1 transfer increases HDL cholesterol but impairs HDL function and accelerates atherosclerosis. Cardiovasc Res 2010;88:376-385.
- Haase CL, Tybjaerg-Hansen A, Grande P, Frikke-Schmidt R. Genetically elevated apolipoprotein A-I, high-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J Clin Endocrinol Metab 2010;95:E500-510.
- Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). Jama 2002;288:49-57.
- Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. Jama 1998;280:605-613.
- Reyes-Soffer G, Ngai CI, Lovato L, Karmally W, Ramakrishnan R, Holleran S, Ginsberg HN. Effect of combination therapy with fenofibrate and simvastatin on postprandial lipemia in the ACCORD lipid trial. Diabetes Care 2013;36:422-428.
- Scott R, O’Brien R, Fulcher G, Pardy C, D’Emden M, Tse D, Taskinen MR, et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009;32:493-498.
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007;357:2109-2122. http://www.ncbi.nlm.nih.gov/pubmed/17984165
- Albers JJ, Slee A, O’Brien KD, Robinson JG, Kashyap ML, Kwiterovich PO, Jr., Xu P, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol 2013;62:1575-1579.
- de Haan W, de Vries-van der Weij J, van der Hoorn JW, Gautier T, van der Hoogt CC, Westerterp M, Romijn JA, et al. Torcetrapib does not reduce atherosclerosis beyond atorvastatin and induces more proinflammatory lesions than atorvastatin. Circulation 2008;117:2515-2522.
- Johannsen TH, Frikke-Schmidt R, Schou J, Nordestgaard BG, Tybjaerg-Hansen A. Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J Am Coll Cardiol 2012;60:2041-2048.
- Li C, Zhang W, Zhou F, Chen C, Zhou L, Li Y, Liu L, et al. Cholesteryl ester transfer protein inhibitors in the treatment of dyslipidemia: a systematic review and meta-analysis. PLoS One 2013;8:e77049.
- Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol 2015;3:507-513.
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 2011;364:127-135.
- Li XM, Tang WH, Mosior MK, Huang Y, Wu Y, Matter W, Gao V, et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 2013;33:1696-1705.
- Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med 2014;371:2383-2393.