
 By B. Corbett Walsh, MD
By B. Corbett Walsh, MD
Peer Reviewed
Case: A 65 year-old man with cirrhosis is admitted to the ICU for severe pneumonia. His hepatic dysfunction is chronically severe, as indicated by a MELD score of 31, and he has a stable thrombocytopenia of 110,000 platelets/ml. Should this patient be on DVT prophylaxis?
MYTH: Patients with severe hepatic dysfunction resulting in a significantly elevated INR are auto-anticoagulated and protected against venous thromboembolic events.
The liver is responsible for a plethora of processes: metabolic (carbohydrate and lipid metabolism, regulating the levels of glucose in the blood), storage (glycogen, triglycerides, iron, vitamins, minerals, etc), catabolic (hormones, proteins, drugs, toxins, alcohol), excretory (bile), and synthetic (certain amino acids, cholesterol, bile, albumin, coagulation factors). Hepatic dysfunction affects all of these processes; however, various models of liver disease severity focus on only a select few. Indeed, the Model for End-stage Liver Disease (MELD) incorporates only the bilirubin, international normalized ratio (INR), and creatinine, with the largest weight given to the INR.
The fundamental model of hemostasis involves the activation of platelets to form a vascular plug, with simultaneous activation of the coagulation cascade (thrombin converting fibrinogen to fibrin) to form a cross-linked fibrin mesh. This coagulation process is elegantly balanced against plasma anticoagulant proteins as well as fibrinolytic factors that may remodel or dissolve existing thrombi. Severe hepatic dysfunction affects every aspect of hemostasis, to be discussed below.
Platelet Function:
Platelets contribute to coagulation in two key ways: first, they interact with vascular endothelial cells to form a hemostatic plug; second, they promote the generation of thrombin. Platelet activation occurs during endothelial damage. Platelets undergo a remarkable shape change to extend pseudopods to form a platelet plug in the damaged vessel. They then adhere to the vessel wall when complexed with von Willebrand Factor (vWF), which is subsequently degraded by ADAMTS13. Platelet activation also brings activated clotting factors to the cell surface to further aid the production of a fibrin mesh [1].
Platelet Function in the context of Cirrhosis:
Platelet function is changed both quantitatively and qualitatively. A large majority of individuals with cirrhosis have thrombocytopenia, thought to be secondary to decreased hepatic synthesis of thrombopoietin, splenic sequestration of platelets, and bone marrow suppression if the etiology of cirrhosis is viral infection (eg, Hepatitis C or HIV). Notably, 75% of cirrhotics have between 100,000 and 150,000 platelets/ml, with 13% having between 50,000 and 100,000 [2]. Platelets above 50,000/ml have not been well correlated with significant bleeding.
In addition to thrombocytopenia, platelet function may also be impaired by concurrent uremia from renal dysfunction. Furthermore, patients with severe hepatic dysfunction have reduced levels of ADAMTS13, thereby increasing active vWF and increasing platelet clot formation.
Coagulation Cascade: the procoagulant players
The liver is the site of synthesis for the majority of the clotting factors, including fibrinogen, thrombin, factor V, VII, IX, X and XI. Not only is there reduced production of procoagulants (with the exception of factor VIII) in liver disease, there is also abnormal processing and glycosylation of factors such as fibrinogen (Table 1), which can affect their function and potentially be prothrombotic. It is well accepted that the summation of these abnormalities culminates in impaired hemostasis. However, thrombin production is surprisingly well preserved in compensated cirrhosis.
Coagulation Cascade: the fibrinolysis players
In addition to synthesizing coagulation factors that promote a hemostatic plug, the liver also produces the inhibitors that counteract procoagulant factors: protein S, protein C, antithrombin, and fibrinolytic factors. It should also be noted that hepatocytes clear circulating vWF. During hepatic dysfunction, the majority of these factors are not produced to the levels seen in healthy individuals (Table 1). Increased tissue plasminogen activator and decreased thrombin-activatable fibrinolysis inhibitor can also be seen [3]. Furthermore, hepatocyte and Kupffer cell dysfunction causes increased levels of circulating vWF [4]. Notably, during some forms of hepatic decompensation, there may also be an associated increase in acute phase reactants like plasminogen activator inhibitor 1 and decreased levels of ADAMTS13, changes that further promote endothelial thrombosis.
Simplified representation of reactions leading to thrombin generation and inhibition [5]Â
Laboratory shortcomings:
Practicing clinicians routinely monitor the prothrombin time (PT), international normalized ratio (INR), and activated partial thromboplasmin time (aPTT), all reflecting the integrity of the coagulation cascade. Tripodi & Mannucci explain the shortcomings of these tests by noting that the INR was initially developed in the 1980s on healthy volunteers to create a standardized test to monitor patients on vitamin K antagonists [6]. High INR values can be misleading in cirrhotic patients with regard to the balance of coagulation, as these tests typically reflect changes in procoagulants, which are reduced in cirrhosis. However, anticoagulant factors are also reduced in patients with severe hepatic dysfunction and often go unmeasured. In addition, the INR is specifically validated for patients stably anticoagulated on vitamin K antagonist. Thus, an INR of 2 in a patient with liver disease is not equivalent to an INR of 2 in a patient that is therapeutically anticoagulated with vitamin K antagonists.
The Myth: Are cirrhotic patients auto-coagulated?
Patients with severe hepatic dysfunction do have impaired regulation and function of their platelets, coagulation, and fibrinolysis pathways. Instead of one process outperforming the other as previously thought, these patients typically remain net even. However, this ‘rebalancing’ makes the system as a whole much more fragile [6] and less capable of appropriately compensating for acute insults such as infection [7], variceal bleeding [8], or uremia [9], potentially causing systemic shifts over the course of hours (Figure 1).
| Table 1. Summary of prohemostatic and Antihemostatic changes in the Different Aspects of Hemostasis in Patients with Severe Hepatic Dysfunction. | ||
| Hemostasis Phase | Prohemostatic Drivers | Antihemostatic Drivers |
| Primary Hemostasis (platelet-vessel wall interactions) | ·       High von Willebrand Factor [4]·       Low ADAMTS [10, 11] | ·       Low platelet count [12] |
| Blood Coagulation (thrombin generation and inhibition) | ·       Low anticoagulant factors: Antithrombin, protein C [13, 14]·       High procoagulant factors: factor VIII [13, 14] | ·       Low procoagulant factors: factors I,II, V, VII, IX, X, XI [13, 14] |
| Fibrinolysis (clot dissolution) | ·       Low plasminogen [15]·       High PAI [15] | ·       High t-Pa [15]·       Low TAFI [16, 17]
·       Low plasmin inhibitor [15] |
| PAI: plasminogen activator inhibitor; TAFI: thrombin-activatable fibrinolysis inhibitor; t-PA tissue plasminogen activatorCBC: measures platelets
PT: measures the extrinsic pathway (uniquely factors VII) PTT: measures the intrinsic pathway (uniquely factors VIII, IX, XI, XII) aa : Measured in both PT & PTT (factors I, II, V, X) |
||
Adapted from [6]
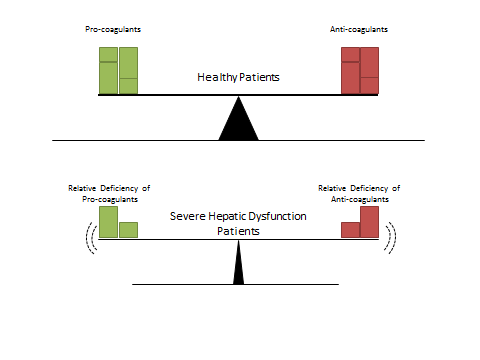
Figure 1. Schematic representation of the coagulation balance found in healthy patients (upper panel) compared with those with severe hepatic dysfunction (bottom panel). Patients in the bottom panel are relatively deficient in both pro- and anti-coagulant factors and therefore have an unstable balance that may tip based on the patient’s current clinical situation.
Adapted from [5]
The myth is therefore Busted. Patients with severe hepatic dysfunction are not auto-anticoagulated against venous thrombotic events as previously thought despite their elevated INR and thrombocytopenia. This previous school of thought was likely due to our inability to measure the overall activity of the coagulation system, which in advanced liver disease becomes rebalanced but with a decreased ability to compensate for acute insults. Such patients like that in the clinical vignette above should be anticoagulated to protect them from venous thromboemboli.
Dr. B. Corbett Walsh is a second year medical resident at NYU Langone Medical Center
Peer reviewed by David Green, MD, hematology, NYU Langone Medicial Center
References
- Tripodi A. Hemostasis in chronic liver disease. J Thromb Haemost. 2006;4(9):2064-2065. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2006.02078.x/pdf
- Afdhal N, McHutchison J, Brown R, Jacobson I, Manns M, Poordad F, et al. Thrombocytopenia associated with chronic liver disease. J Hepatol. 2008;48(6):1000-1007. https://www.ncbi.nlm.nih.gov/pubmed/18433919
- Van Thiel DH, George M, Fareed J. Low levels of thrombin activatable fibrinolysis inhibitor (TAFI) in patients with chronic liver disease. Thromb Haemost. 2001;85(4):667-670.
- Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44(1):53-61.
- Tripodi A. Hemostasis abnormalities in cirrhosis. Curr Opin Hematol. 2015;22(5):406-412. https://www.ncbi.nlm.nih.gov/pubmed/26203733
- Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011;365(2):147-156. https://www.ncbi.nlm.nih.gov/pubmed/21751907
- Ferro D, Basili S, Lattuada A, Mantovani B, Bellomo A, Mannucci PM, et al. Systemic clotting activation by low-grade endotoxaemia in liver cirrhosis: a potential role for endothelial procoagulant activation. Ital J Gastroenterol Hepatol. 1997;29(5):434-440. https://moh-it.pure.elsevier.com/en/publications/systemic-clotting-activation-by-low-grade-endotoxaemia-in-liver-c
- Goulis J, Armonis A, Patch D, Sabin C, Greenslade L, Burroughs AK. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal hemorrhage. Hepatology. 1998;27(5):1207-1212.
- Sagripanti A, Cupisti A, Baicchi U, Ferdeghini M, Morelli E, Barsotti G. Plasma parameters of the prothrombotic state in chronic uremia. Nephron. 1993;63(3):273-278.
- Lodge JP, Jonas S, Jones RM, Olausson M, Mir-Pallardo J, Soefelt S, et al. Efficacy and safety of repeated perioperative doses of recombinant factor VIIa in liver transplantation. Liver Transpl. 2005;11(8):973-979.
- Feys HB, Canciani MT, Peyvandi F, Deckmyn H, Vanhoorelbeke K, Mannucci PM. ADAMTS13 activity to antigen ratio in physiological and pathological conditions associated with an increased risk of thrombosis. Br J Haematol. 2007;138(4):534-540.
- Giannini EG, Savarino V. Thrombocytopenia in liver disease. Curr Opin Hematol. 2008;15(5):473-480.
- Tripodi A, Primignani M, Chantarangkul V, Dell’Era A, Clerici M, de Franchis R, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology. 2009;137(6):2105-2111.
- Tripodi A, Primignani M, Lemma L, Chantarangkul V, Dell’Era A, Iannuzzi F, et al. Detection of the imbalance of procoagulant versus anticoagulant factors in cirrhosis by a simple laboratory method. Hepatology. 2010;52(1):249-255.
- Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, Reddy KR, et al. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology. 2006;44(4):1039-1046.
- Lisman T, Leebeek FW, Mosnier LO, Bouma BN, Meijers JC, Janssen HL, et al. Thrombin-activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology. 2001;121(1):131-139.
- Colucci M, Binetti BM, Branca MG, Clerici C, Morelli A, Semeraro N, et al. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology. 2003;38(1):230-237.