
 By Zachary Henig
By Zachary Henig
Peer Reviewed
Atherosclerosis is the primary risk factor for cardiovascular disease, the leading cause of mortality worldwide. To understand the pathophysiology of atherosclerosis, we turn to advances made in molecular biology. Understanding the mechanisms underlying this process enables a more nuanced approach to the strategies used to affect the natural course of the disease.
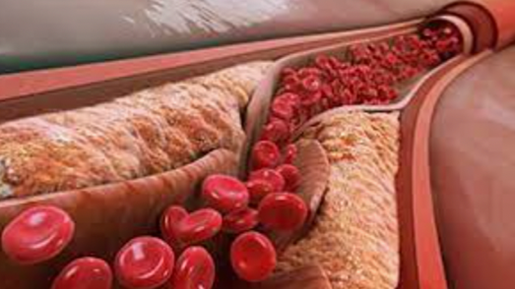
Atherosclerosis is broadly characterized by the accumulation of lipids, fibrous elements, and calcification within large arteries.1 The vascular endothelium is the first barrier to all substances circulating in the bloodstream. The endothelium responds to its immediate environment, such as changes in mechanical stress from altered blood pressure and changes in concentrations of metabolites. Of these factors, the mechanical forces that reduce wall shear stress (WSS) on the endothelium, such as turbulent flow generated at vessel bifurcations and highly curved vasculature, confer the highest risk for atherosclerosis.2,3 Reduced WSS promotes endothelial dysfunction and subsequent low-density lipoprotein (LDL) infiltration, which constitutes the first step in atheroma formation.1
Once endothelial dysfunction has initiated, LDL retention in the tunica intima4,5 promotes the endothelial cells to recruit monocytes to the area. These LDL particles are then captured by monocytes and vascular smooth muscle cells (VSMCs) to promote foam cell formation.6 Simultaneously, several inflammatory processes are activated, leading to fatty streak formation characterized by the accumulation of lipids within monocytes and VSMCs and in the extracellular milieu.7
The next step in this pathogenic process is fibrous plaque development. Here, the fatty streak transitions to intimal growing, characterized by the formation of a necrotic core that is lipid-rich and cell-free. This necrotic core is covered by a fibrous cap, constituting advanced atherosclerosis.8 Once this stage is reached, plaque regression is unlikely to occur.9
As plaques grow, they experience increased stress on their shoulders, making the lesions prone to rupture.10 A rupture-prone plaque is characterized by a large necrotic core, a thin fibrous cap, and an increased inflammatory response.11 Inflammation, which is critical for all processes involved with atheroma plaque formation, promotes the final instability of the fibrous cap and eventual plaque rupture.10
There is increasing interest and understanding of the role of long non-coding RNAs (lncRNAs) as regulators in the atherosclerotic march. While once thought to be “junk of the genome,†there is an appreciation that they may influence disease-associated genes.12 lncRNAs are sequences longer than 200 nucleotides that exhibit mRNA-like features and influence transcriptional and translational processes. For example, a lncRNA called metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been implicated in controlling endothelial cell inflammation.13,14 MALAT1, whose expression is increased during hypoxia and oxidative stress, is thought to induce the expression of autophagy-related genes and, subsequently, endothelial cell survival.15 Without delving into the molecular mechanisms of these effects, there is ample evidence from in-vitro and in-vivo rodent studies to support these findings.12-15 LncRNAs have been studied in many domains related to atherosclerotic progression, including cardiomyocytes, endothelial cells, smooth muscle cells, monocyte and macrophage function, and lipids.12 While further studies are needed to understand the translational value of lncRNAs, it is possible that one day the medical-scientific community will be able to target them or harness their diagnostic potential in the fight against cardiovascular morbidity and mortality. Furthermore, lncRNAs represent just one area of enhanced research efforts; other areas of pathophysiological mechanisms under investigation include the roles of inflammation, microRNAs, and the gut microbiota.
In sum, the evolution from healthy vascular endothelium to diseased atherosclerotic tissue follows a progression that has largely been elucidated. However, questions remain, and further research is ongoing, particularly as it pertains to novel biologic molecules and pathways such as lncRNAs, microRNAs, and the roles of inflammation and the gut microbiota. In time, these novel research avenues will give way to a more nuanced understanding of atherosclerosis and uncover more questions about the disease that remain hidden from us today. As clinicians, this sort of scientific progress is what enables us to help our patients.
Zachary Henig is a 3rd year medical student at NYU Grossman School of Medicine
Reviewed by Michael Tanner, MD, Associate editor, Clinical Correlations
Image courtesy of Wikimedia Commons, source:Â Manu5, CC BY-SA 4.0 https://creativecommons.org/licenses/by-sa/4.0
ReferencesÂ
- Jebari-Benslaiman S, Galicia-Garcia U, Larrea-Sebal A, et al. Pathophysiology of atherosclerosis. Int J Mol Sci. 2022;23(6):3346. https://doi.org/10.3390/ijms23063346
- Roger VL, Weston SA, Killian JM, et al. Time trends in the prevalence of atherosclerosis: a population-based autopsy study. Am J Med. 2001;110(4):267-273. https://doi.org/10.1016/s0002-9343(00)00709-9
- Targonski P, Jacobsen SJ, Weston SA, et al. Referral to autopsy: effect of antemortem cardiovascular disease: a population-based study in Olmsted County, Minnesota. Ann Epidemiol. 2001;11(4):264-270. https://doi.org/10.1016/s1047-2797(00)00220-9
- Hermida N, Balligand JL. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: the role of statins. Antioxid Redox Signal. 2014;20(8):1216-1237. https://doi.org/10.1089/ars.2013.5537
- Mundi S, Massaro M, Scoditti E, et al. Endothelial permeability, LDL deposition, and cardiovascular risk factors–a review. Cardiovasc Res. 2018;114(1):35-52. https://doi.org/10.1093/cvr/cvx226
- Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245-252. https://doi.org/10.1016/j.cca.2013.06.006
- Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89(5):2462-2478. https://doi.org/10.1161/01.cir.89.5.2462
- Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233-241. https://doi.org/10.1038/35025203
- Fisher EA, Feig JE, Hewing B, Hazen SL, Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32(12): 2813-2820. https://doi.org/10.1161/ATVBAHA.112.300133
- Slager CJ, Wentzel JJ, Gijsen FJ, et al. The role of shear stress in the generation of rupture-prone vulnerable plaques. Nat Clin Pract Cardiovasc Med. 2005;2(8):401-407. https://doi.org/10.1038/ncpcardio0274
- Boudoulas KD, Stefanadis C, Boudoulas H. The role of interventional cardiology to our understanding of basic mechanisms related to coronary atherosclerosis: “Thinking outside the box”. Hellenic J Cardiol. 2017;58(2):110-114. https://doi.org/10.1016/j.hjc.2016.10.002
- Haemmig S, Simion V, Feinberg MW. Long non-coding RNAs in vascular inflammation. Front Cardiovasc Med. 2018;5:22. https://doi.org/10.3389/fcvm.2018.00022
- Michalik, KM, You X, Manavski Y, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389-1397. https://doi.org/10.1161/CIRCRESAHA.114.303265
- Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015; 19(6):1418-1425. https://doi.org/10.1111/jcmm.12576 6
- Li Z, Li J, Tang N. Long noncoding RNA Malat1 is a potent autophagy inducer protecting brain microvascular endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury by sponging miR-26b and upregulating ULK2 expression. Neuroscience. 2017;354:1-10. https://doi.org/10.1016/j.neuroscience.2017.04.017