
 By Leonard Naymagon, MD
By Leonard Naymagon, MD
Peer Reviewed
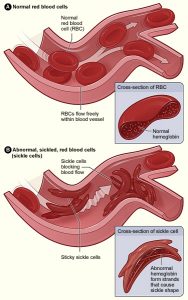
Vaso-occlusive crisis (VOC), or pain crisis, is the most common clinical manifestation of sickle cell disease (SCD) and is responsible for the majority of emergency department (ED) visits and inpatient hospitalizations among sickle cell patients [1]. A retrospective analysis of over 100,000 in-hospital encounters for VOC in 2005 and 2006 demonstrated 30-day and 14-day rehospitalization rates of 33.4% and 22.1% respectively [1]. These findings were most pronounced among 18- to 30-year-olds, with 41.1% rehospitalized within 30 days and 28.4% within 14 days. Among publicly insured patients of this age group, total in-hospital encounters (either ED or inpatient) numbered 4.8 per patient per year. Studies at specific high-volume urban medical centers have shown one-month readmission rates for VOC as high as 50% [2]. For many years, and among many physicians, frequent ED visits and hospitalizations were thought to reflect the nature of VOC as an obligatory, often inescapable, product of SCD. More cynically, high acutecare utilization rates among this patient population have often been anecdotally associated with malingering behaviors, such as drug-seeking. Increasingly, however, frequent ED visits, hospitalizations, and re-admissions are coming to be understood as symptoms of a suboptimal model of care, which can fail SCD patients on numerous fronts [1], [3]. Recent initiatives at many medical centers have demonstrated that the most effective improvements to the current model likely lie in easing and expanding access to specialized primary care and embracing the notion of patient-centered medical homes [4], [5], [6], [7]. Such shifts in philosophy may significantly improve quality of life among SCD patients while significantly reducing costs associated with frequent hospital-based encounters.
Patients with SCD should have regular follow-up with a specialized clinician and treatment team as part of a comprehensive healthcare maintenance program [6]. Given the relatively extensive screening and prophylaxis guidelines, the wide array of potential complications, and the intensity of their baseline analgesic requirements, SCD patients are typically best served by a primary physician who has some degree of subspecialization in their care, most often a hematologist. Additionally, because they generally require regular follow-up with other specialists (including ophthalmologists, vascular surgeons, pain management physicians, psychiatrists, and social workers, among others), they are best served within patient-centered medical homes (PCMHs) [4], [7]. SCD patients will often be burdened with numerous appointments, which, if localized at disparate sites with little if any cross-communication, will result in substandard care, patient frustration, and eventual lack of compliance [7]. The PCMH model in SCD is most robust among the pediatric population [6], [7]. As young adults age out of their pediatric medical homes, they are often left to scramble for adequate adult follow-up, and  the follow-up they do find is often less comprehensive than that to which they were previously accustomed [6],[7]. During this transition in care, SCD patients tend to be at their most vulnerable It is likely for this reason that ED usage, hospitalization, and readmission rates are greatest among this age group [1],[6],[7].
Adequate outpatient follow-up remains a major issue among the adult SCD population. A study conducted at Boston University Medical Center demonstrated that at baseline, only 46% of patients admitted for VOC were discharged with a hematology appointment within 28 days, only 19% of VOC discharges actually followed up with a hematologist within 28 days, and the rate of re-presentation to the ED within 30 days of hospital discharge was 44% [8]. Barriers to the establishment of outpatient follow-up included a limited availability of hematology appointments (only 2 hematologists at the medical center routinely saw patients with SCD), a cumbersome process for the scheduling of appointments, an inability to book appointments during clinic off-hours, and patient socioeconomic factors. An initiative at Virginia Commonwealth University which increased outpatient visits among SCD patients by approximately 50% was shown to coincide with an approximate 90% reduction in ED visits, 50% reduction in hospital admissions, and 60% reduction in hospital days [4]. Much of this improvement was achieved by targeting those patients with particularly high ED utilization rates.
The above institutional experiences highlight the importance of discharge planning with respect to SCD patients. Ensuring adequate follow-up at the time of discharge is a simple way to prevent future admissions and ED visits. However, anticipating discharge for a VOC patient is not often easy, as the length of course is difficult to predict, and patients may either improve or deteriorate rapidly. Furthermore, the conclusion of a VOC is an inexact point in time, colored by subjectivity on the part of both patient and clinician. Discharge from the ED or hospital often does not indicate the end of the crisis. It is “an arbitrary point that depends on several factors that are not based on the individual status of the patient but subscribes to insurance coverage, hospital policy, and the attitude of the providers†[9]. Many clinicians have come to associate numerous prolonged hospital stays as signs of malingering or drug-seeking. Although this may sometimes be the case, such biases may promote premature discharges, which are prone to rapid “bounce-back.†In VOC, more so than in any other diagnosis, provider subjectivity and bias plays a major role in determining the hospital course and discharge circumstances. In one study, 86% of clinicians in academic centers did not accept that patient self-report is the best metric for pain among patients with SCD [10]. In another, 50% of surveyed emergency physicians believed that patients presenting with VOC were abusing opioids [11]. Patients who are routinely perceived to be drug-seeking are less likely to be afforded good outpatient follow-up, as the severity of their condition and the probability of their compliance are often underestimated. For these same reasons, during those instances that close follow-up is arranged, they are unlikely to be discharged with sufficient pain medications to last through their follow-up appointment, predisposing them to re-presentation when their medications run out.
The subjectivity involved in treating VOC may be mitigated to a degree by the implementation of “care plans.†Such plans may be established by a patient’s outpatient providers or prior to discharge following a hospitalization and should instruct both the patient and potential future providers about appropriate management of mild, moderate, and severe pain with clearly defined thresholds for the use of opioids and presentation to the ED [12]. Such plans help guide patients through home pain-management strategies and, when available to acute care providers during the patient’s next ED visit or hospital admission, help provide collateral regarding the patient’s typical analgesic requirements, thus limiting at least one source of subjectivity and bias. Studies have shown that the establishment of personalized care plans for VOC have reduced hospitalization rates, length of stay, and readmission rates [5], [13].
Few interventions have been shown to decrease the incidence of VOC among SCD patients. Of those that exist, hydroxyurea (HU) is the most validated and likely the most effective. HU has been shown to reduce the median rate of VOC by nearly 50% [14]. It has also been shown to reduce mortality, reduce the incidence of acute chest syndrome, reduce transfusion requirement, be well tolerated, and be cost effective [14], [15], [16]. HU is indicated in all adult SCD patients with a history of frequent VOCs or complications from VOC. However, despite its significant benefits and broad indications, it remains woefully underused. In a retrospective trial including 677 patients who experienced at least 3 VOCs during the course of 1 year, only 15% of patients filled a prescription for HU within 3 months of their third encounter [17]. Initiation of HU is typically pursued on an outpatient basis, and maintenance requires frequent outpatient follow-up for regular laboratory evaluation. HU underuse is likely a symptom of inadequate outpatient access and follow-up. [17]. Any VOC patients not actively taking HU should be asked why. Absolute contraindications are few but include pregnancy and development of cytopenias despite dose reduction. An effort should be made to start all eligible SCD patients on HU. This effort is best pursued by discussing potential risks and benefits with non-adherent patients at every opportunity and by ensuring adequate outpatient follow-up.
A well-established PCMH can prevent ED visits, admissions, and readmissions among SCD patients, but it cannot eliminate instances of acute, severe VOC. Increasingly, however, healthcare systems are trying to shift the management of VOC into the domain of the PCMH [18], [19]. In 2008, Johns Hopkins Hospital opened a dedicated acute care facility for adults presenting with VOC [18]. This sickle cell infusion center (SCIC) operates as a means of providing short-term intravenous analgesia outside of the ED setting, increasing the ease and rapidity with which SCD patient can receive urgent care. The SCIC offers on-site ancillary services, including social work and metal health, thus acting as a PCMH in the acute setting. From 2008 to 2011, the SCIC saw 3,874 patient visits compared with 3,408 visits to the ED during the same period [18]. The opening of the SCIC coincided with a significant reduction in ED visits for VOC in the city of Baltimore. There was a significant decrease in readmissions over time for the entire Baltimore Metro area with the likelihood of readmission decreasing by 7%. Furthermore, 85% of visits to the SCIC resulted in patients being sent home, and the hospital admission rate at the SCIC was 42% that of the ED. Cost benefit analysis demonstrated that the SCIC yielded a 7.6% cost savings during the first year of its operation [19]. The Johns Hopkins SCIC provides a model for how PCMHs can improve outcomes and quality of life among SCD patients while decreasing the costs related to recurrent ED visits and hospitalizations.
Dr. Leonard Naymagon is a 2nd year resident at NYU Langone Health
Peer reviewed by David Green, MD, PhD, Clinical Assistant Professor, Division of Hematology/Oncology, Department of Medicine, NYU Langone Health
Image courtesy of Wikimedia Commons
References:
- Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303(13):1288-94.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol. 2005;79(1):17-25.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014; 312:1033.
- Roberts, John D. “Caring for Adults with Sickle Cell Disease at Two Institutions.” Yale University Medical Center, New Haven. 13 June 2014. Lecture.
- Krishnamurti L, Smith-Packard B, Gupta A, et al. Impact of individualized pain plan on the emergency management of children with sickle cell disease. Pediatr Blood Cancer 2014; 61:1747.
- Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-Care Indicators for Children With Sickle Cell Disease. Pediatrics 2011; 128:484.
- Raphael JL, Oyeku SO. Sickle cell disease pain management and the medical home. Hematology Am Soc Hematol Educ Program. 2013;2013:433-8.
- Slade J, Wolfgang T, Kavanagh P (2014, January). Improving Outpatient Follow-Up for Patients with Sickle Cell Disease. Poster session presented at Boston University School of Medicine Research Day, Boston, MA.
- Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647-56.
- Labbé E, Herbert D, Haynes J. Physicians’ attitude and practices in sickle cell disease pain management. J Palliat Care 2005; 21:246.
- Shapiro BS, Benjamin LJ, Payne R, Heidrich G. Sickle cell-related pain: perceptions of medical practitioners. J Pain Symptom Manage 1997; 14:168.
- Matthie N, Jenerette C. Sickle cell disease in adults: developing an appropriate care plan. Clin J Oncol Nurs. 2015;19(5):562-7.
- Liles AE, Kirsch J, Gilchrist M, et al. Hospitalist management of vaso-occlusive pain crisis in patients with sickle cell disease using a pathway of care. Hosp Pract (1995) 2014; 42:70.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995; 332:1317.
- Davies S, Olujohungbe A. Hydroxyurea for sickle cell disease. Cochrane Database Syst Rev 2001;(2):CD002202.
- Moore RD, Charache S, Terrin ML, et al. Cost-effectiveness of hydroxyurea in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Am J Hematol 2000; 64:26.
- Stettler N, McKiernan CM, Melin CQ, et al. Proportion of adults with sickle cell anemia and pain crises receiving hydroxyurea. JAMA 2015; 313:1671.
- Lanzkron S, Carroll CP, Hill P, et al. Impact of a dedicated infusion clinic for acute management of adults with sickle cell pain crisis. Am J Hematol. 2015;90(5):376-80.
- Chappidi M, Alfonso YN, Biahai D,et al. Cost Benefit Analysis Of a Sickle Cell Infusion Center For The Treatment Of Vaso-Occlusive Crises. Blood. 2013; 122 (21).