 By Chio Yokose, MD
By Chio Yokose, MD
Peer Reviewed
Even in this era of modern medicine, bacterial meningitis remains a widely feared diagnosis in both resource-rich and -poor settings worldwide. Bacterial meningitis is among the ten most …

By Chio Yokose, MD
Peer Reviewed
Even in this era of modern medicine, bacterial meningitis remains a widely feared diagnosis in both resource-rich and -poor settings worldwide. Bacterial meningitis is among the ten most …
 By Gabriel Campion
By Gabriel Campion
Peer Reviewed
For over a century, neckties have been a staple accessory in the wardrobe of the American professional man. Although white-collar dress codes have trended toward a more casual …
 By Amy Shen Tang, MD
By Amy Shen Tang, MD
Peer Reviewed
“I would pay you if you took it away from me. I’d try to buy it back,†said Irving Kahn, the late Wall Street investment advisor …
 By Jenna Tarasoff
By Jenna Tarasoff
Peer Reviewed
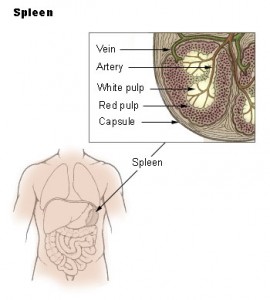
A 65-year-old African woman presents with two months of fevers and 25-pound weight loss along with a month of nausea and retching, accompanied by left-sided abdominal pain. …
 By Amar Parikh, MDÂ
By Amar Parikh, MDÂ
Peer Reviewed
Amidst the global panic over the recent Ebola outbreak, another well-known pathogen that has been devastating the world for decades continues to smolder—the human immunodeficiency virus (HIV). …
 By Lauren Strazzulla
By Lauren Strazzulla
Current FDA guidelines for the use of metformin stipulate that it not be prescribed to those with an elevated creatinine (at or above 1.5 mg/dL for men and 1.4 mg/dL for women). It is …
 By Shimwoo Lee
By Shimwoo Lee
Peer Reviewed
Case: A 31-year-old man with poorly controlled type 2 diabetes was hospitalized for community-acquired pneumonia. His home medications included esomeprazole. When asked why he was receiving this …
 By Robin Guo, MD
By Robin Guo, MD
Peer ReviewedÂ
Beta-blockers were one of the first modern medications used for the treatment of blood pressure. Before 1950, treatment options for hypertension were limited. The alphabet …