 By Jenna Conway, MD
By Jenna Conway, MD
Peer Reviewed
IntroductionÂ
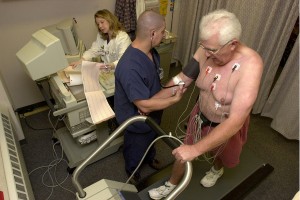
A 58-year-old man presents with worsening dyspnea and nonproductive cough for five days. Significant history includes a recent hospitalization for congestive heart failure. He is …

By Jenna Conway, MD
Peer Reviewed
IntroductionÂ
A 58-year-old man presents with worsening dyspnea and nonproductive cough for five days. Significant history includes a recent hospitalization for congestive heart failure. He is …
 By Amar Parikh, MD
By Amar Parikh, MD
Peer Reviewed
Welcome to Gamechangers, a series that takes a critical look at the latest in medical literature to answer one important question: would the results of this article change my …
 By Iulia Giuroiu, MD
By Iulia Giuroiu, MD
Peer Reviewed
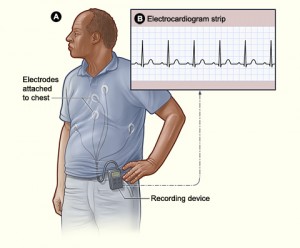
A 70-year-old woman with hypertension, early dementia, and non-specific chest pain of unclear etiology presents with recurrent left-sided chest pain. Unfortunately, she is a poor historian; it …
 By Kerrilynn Carney, MD
By Kerrilynn Carney, MD
Peer Reviewed
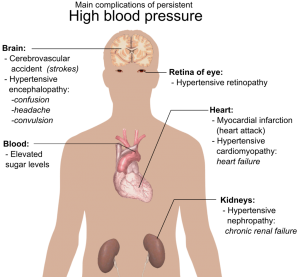
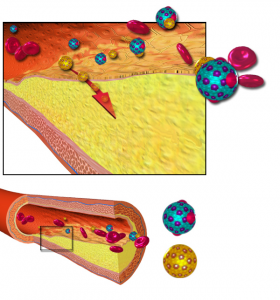
Coronary heart disease remains the leading cause of death globally despite the use of statin therapy. Although major statin studies suggest an average 31% reduction in relative …
 By Steven Bolger
By Steven Bolger
Peer Reviewed
Omega-3 fatty acids were first identified as a potential agent to prevent and treat cardiovascular disease through several epidemiologic studies of the Greenlandic Inuit in the 1970s suggesting …
 Cindy Fei, MD
Cindy Fei, MD
Peer Reviewed
A 63-year-old woman with hypertension presents to your clinic for routine follow-up. She came across an online article regarding C-reactive protein and its purported link to heart disease, and she asks you whether she should be tested …
 By Shannon Chiu, MD
By Shannon Chiu, MD
Peer Reviewed
The annual incidence of infective endocarditis (IE) is estimated to be 3 to 9 cases per 100,000 persons in developed countries [1-2]. Neurologic complications are the most …
 By Anjali Varma Desai, MD
By Anjali Varma Desai, MD
Peer Reviewed
Mr. Q is a 55-year-old male smoker who presents with recurrent chest pain in the mornings over the past several months. The patient reports being awakened from sleep at approximately 5:00 a.m. each …