

By Dr. Michael Tanner
The author, at age 14 in 1968, with pennies on his eyes, wearing a Napoleonic military jacket fashionable at the time.
Albums clockwise from top left:
After Bathing at Baxter’s (Jefferson Airplane),
Blonde on Blonde (Bob Dylan),
Disraeli Gears (Cream),
Magical Mystery Tour …
 By Carolyn Akers
By Carolyn Akers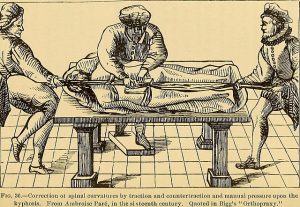 By: Michael Moore
By: Michael Moore By Daniel Gratch, MD
By Daniel Gratch, MD By Kevin Rezzadeh
By Kevin Rezzadeh Join us in this episode as we question everything you ever thought you knew about… urinary tract infections (UTI) and delirium. || By Steven R. Liu MD, …
Join us in this episode as we question everything you ever thought you knew about… urinary tract infections (UTI) and delirium. || By Steven R. Liu MD, …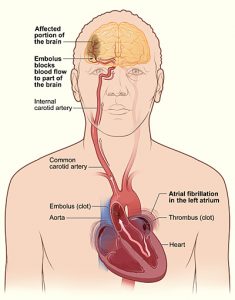 By Dixon Yang, MD
By Dixon Yang, MD