 By Hadley Greenwood
By Hadley Greenwood
Peer Reviewed
Medications originally developed as agents to treat type 2 diabetes have been making headlines for their use as weight loss drugs, even in individuals without diabetes. The medical community …

By Hadley Greenwood
Peer Reviewed
Medications originally developed as agents to treat type 2 diabetes have been making headlines for their use as weight loss drugs, even in individuals without diabetes. The medical community …
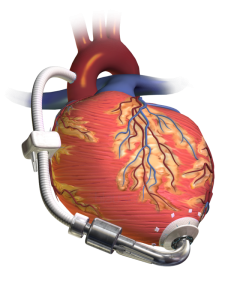 By Joshua Novack, MD
By Joshua Novack, MD
Peer Reviewed
Case: 74 year old male with a history of heart failure with reduced ejection fraction (EF 20%) diagnosed 10 years ago comes in with subacute progressive lower extremity edema, …
 By Luke Bonanni
By Luke Bonanni
Peer Reviewed
Theobroma, literally “food of gods†in Greek, is an apt description of chocolate. Made from the fermented seeds of the Theobroma cacao tree, chocolate is an immensely popular …
 By Akshay N. Pulavarty MPH
By Akshay N. Pulavarty MPH
Peer Reviewed
A quick look into the medicine cabinet of anyone sixty or older will likely reveal a statin. Primary prevention with high-intensity statins has substantially reduced the …
 By Mahip Grewal
By Mahip Grewal
Peer Reviewed
Nonalcoholic fatty liver disease (NAFLD) is estimated to affect 25% of the world’s population.1 NAFLD is a spectrum of disease ranging from nonalcoholic fatty liver (NAFL), which is …
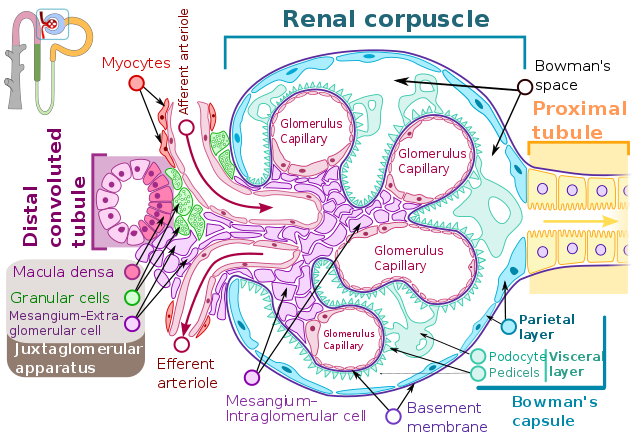 By Logan Groneck
By Logan Groneck
Peer Reviewed
Your next patient is very familiar. The management of her chronic diseases–heart failure (HF) and chronic kidney disease (CKD)—has been optimized. Like the 6.2 million other Americans with …
 By Joachner Philippe
By Joachner Philippe
Peer ReviewedÂ
Ms. G was a 44-year-old African American woman with a history of sarcoidosis presenting to her outpatient pulmonologist/primary care physician for follow-up. Insurance complications had forced her to …
 By Emily Mills
By Emily Mills
Peer Reviewed
While on my medical school Pediatrics rotation I learned of an important developmental milestone—the “why†phase. During this time, the inquisitive three-year-old bombards their parents with an infinite …