 By Nicolas Gillingham
By Nicolas Gillingham
Peer Reviewed
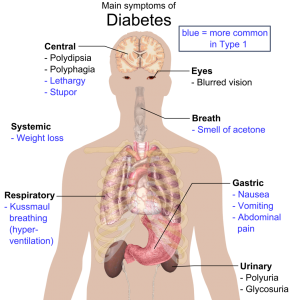
Over 30 million Americans—9.4% of the population—live with diabetes, six million of whom are at least partially dependent on exogenous insulin.[1] Insulin can be self-administered …

By Nicolas Gillingham
Peer Reviewed
Over 30 million Americans—9.4% of the population—live with diabetes, six million of whom are at least partially dependent on exogenous insulin.[1] Insulin can be self-administered …
 By Allison Harrington, MD
By Allison Harrington, MD
Peer Reviewed
Learning Objectives:
1) When should diabetic myonecrosis be suspected?
2) What are the diagnostic criteria for diabetic myonecrosis? What is the pathophysiology?
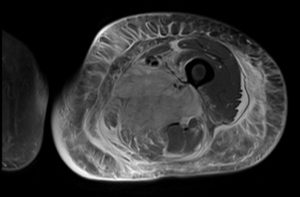
3) What is the management of diabetic myonecrosis? How can it …
 By Alexa Yuen
By Alexa Yuen
Peer ReviewedÂ
According to the CDC, there are 22 million people with an established diagnosis of diabetes mellitus in the United States, but more frightening is that …
 By Johanna Hase, MD
By Johanna Hase, MD
Peer Reviewed
Welcome to Gamechangers, a series that takes a critical look at the latest in medical literature to answer one important question: would the results …
 Samantha Kass Newman, MD
Samantha Kass Newman, MD
Peer Reviewed
You can get a Botox injection almost anywhere these days. Internists, dermatologists, and even gynecologists have capitalized on an aging group of baby boomers who aren’t fans …
 By Amy Shen Tang, MD
By Amy Shen Tang, MD
Peer Reviewed
“I would pay you if you took it away from me. I’d try to buy it back,†said Irving Kahn, the late Wall Street investment advisor …
 By Lauren Strazzulla
By Lauren Strazzulla
Current FDA guidelines for the use of metformin stipulate that it not be prescribed to those with an elevated creatinine (at or above 1.5 mg/dL for men and 1.4 mg/dL for women). It is …
 By Shilpa Mukunda, MD
By Shilpa Mukunda, MD
Peer Reviewed
On my first day on inpatient medicine at the VA Hospital, Mr. P came in with an oozing foot ulcer. Mr. P, a 60-year-old man with …