 By Ipsita Subudhi
By Ipsita Subudhi
Peer Reviewed
When a patient comes in with skin plaques, bumps, and what might be a rash, the natural reaction might be to assume that these issues only lie …

By Ipsita Subudhi
Peer Reviewed
When a patient comes in with skin plaques, bumps, and what might be a rash, the natural reaction might be to assume that these issues only lie …
 By Matthew Auda
By Matthew Auda
Peer ReviewedÂ
Let’s begin with a case. The patient is a 67-year-old female with a past medical history of hypothyroidism referred to your Cardiology Clinic by her primary care physician for cardiovascular …
 By Zachary Henig
By Zachary Henig
Peer Reviewed
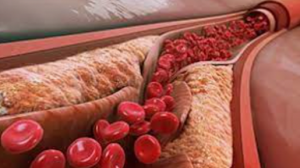
Atherosclerosis is the primary risk factor for cardiovascular disease, the leading cause of mortality worldwide. To understand the pathophysiology of atherosclerosis, we turn to advances made in molecular …
 By Pamela Boodram, MD
By Pamela Boodram, MD
Peer Reviewed
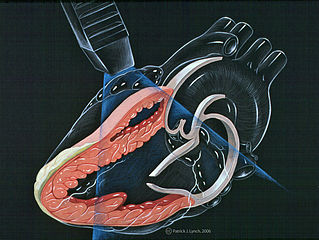
A 68-year-old woman with a history of hypertension and well controlled type 2 diabetes presents to the ED with five days of progressively worsening dyspnea on exertion, …
 By Lily Cao
By Lily Cao
Peer ReviewedÂ
A quick web search would suggest countless reasons to take fish oil, a supplement that Americans have fallen in love with. In …
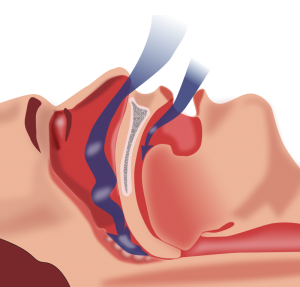 By Gregory Rubinfeld, MD
By Gregory Rubinfeld, MD
Peer Reviewed
Obstructive sleep apnea (OSA) is an increasingly prevalent disorder that has well described associations with cardiovascular disease. OSA affects approximately 20–30% of males and 10–15% of females in North America.1-3 In addition to male gender, other risk …
 By David Pineles, MD
By David Pineles, MD
Peer Reviewed
With the implementation of the ACC/AHA guidelines on assessment of cardiovascular risk, HMG-CoA reductase inhibitors, commonly known as statins, will become one of the most prescribed medications in history [1]. Dr. John Ioadnnidis, in an article published …
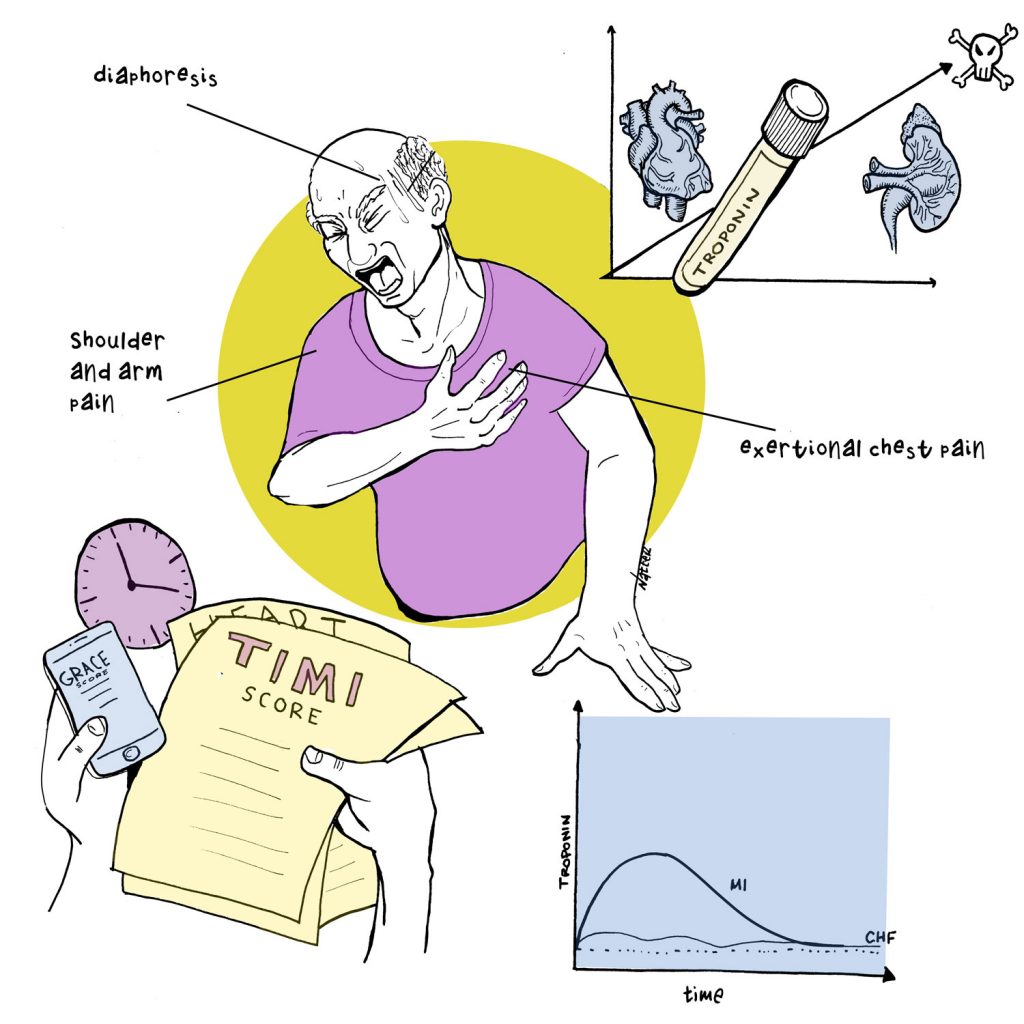 By David Rhee MD, Greg Katz MD, Marty Fried MD, Shreya P. Trivedi MD || Illustration by Michael Natter MD || …
By David Rhee MD, Greg Katz MD, Marty Fried MD, Shreya P. Trivedi MD || Illustration by Michael Natter MD || …