
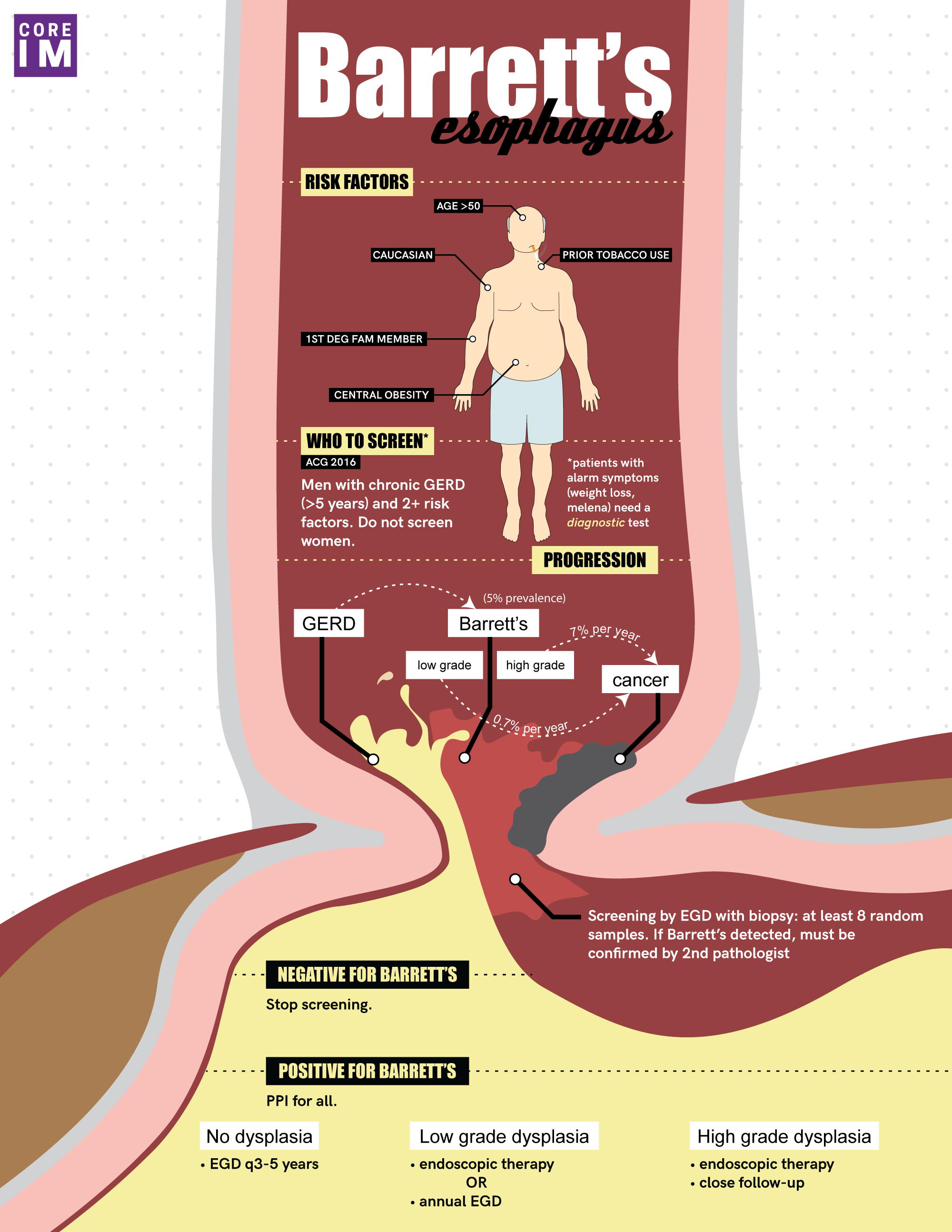
By Vishal Shah MD, Milna Rufin MD, Marty Fried MD, Shreya P. Trivedi MD || Illustration by Amy Ou MD || Audio Editing by Harit Shah. Quiz yourself on the 5 Pearls we will be covering:
- What is Barrett’s esophagus? (4:12)
- Who do we screen for Barrett’s, and why? (8:41)
- How do we …
 By Rebecca Lazarus, MD
By Rebecca Lazarus, MD By Kevin Rezzadeh
By Kevin Rezzadeh By David Pineles, MD
By David Pineles, MD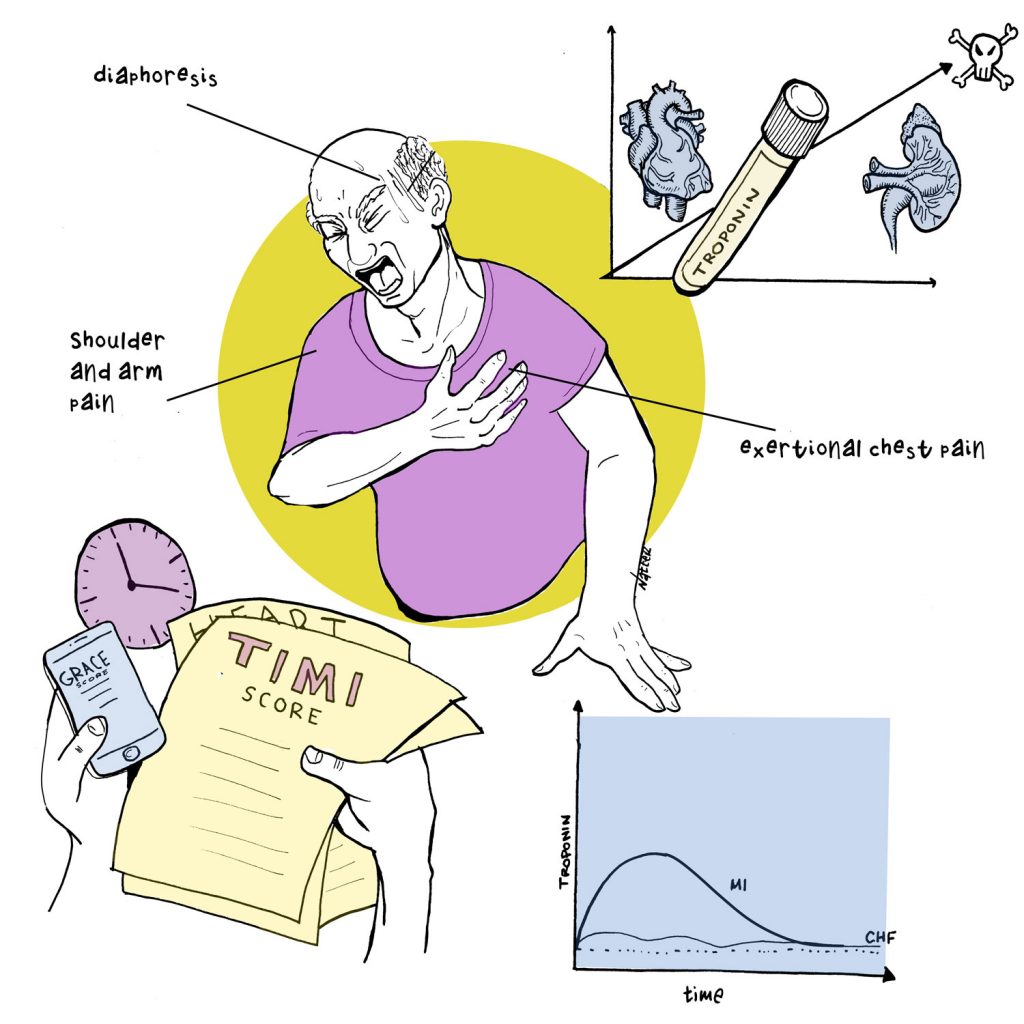 By David Rhee MD, Greg Katz MD, Marty Fried MD, Shreya P. Trivedi MD || Illustration by Michael Natter MD || …
By David Rhee MD, Greg Katz MD, Marty Fried MD, Shreya P. Trivedi MD || Illustration by Michael Natter MD || … By Jessica K Qiu
By Jessica K Qiu By Matthew Vorsanger MD, David Kudlowitz MD, and Patrick Cocks MD
By Matthew Vorsanger MD, David Kudlowitz MD, and Patrick Cocks MD