
By Jamie Oliver
Peer Reviewed
Colorectal cancer (CRC) is the second most common cause of cancer death in the US.[1]Almost all CRC develops from precancerous polyps, and with appropriate colonoscopy screening most …

By Jamie Oliver
Peer Reviewed
Colorectal cancer (CRC) is the second most common cause of cancer death in the US.[1]Almost all CRC develops from precancerous polyps, and with appropriate colonoscopy screening most …
 By Allison Guttmann, MD
By Allison Guttmann, MD
Peer Reviewed
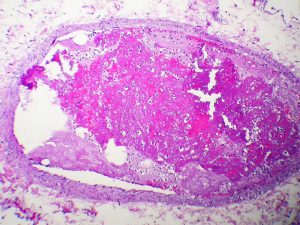
A middle-aged woman presents to the Emergency Department with 1 week of calf-pain and swelling. A venous duplex ultrasound reveals a non-compressible popliteal vein suggestive of …
 By Gregory Rubinfeld, MD
By Gregory Rubinfeld, MD
Peer Reviewed
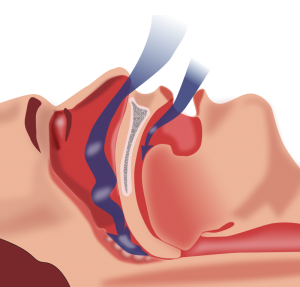
Obstructive sleep apnea (OSA) is an increasingly prevalent disorder that has well described associations with cardiovascular disease. OSA affects approximately 20–30% of males and 10–15% of females in North America.1-3 In addition to male gender, other risk …
 By Rebecca Lazarus, MD
By Rebecca Lazarus, MD
Peer Reviewed
The concept of lactate is frequently on the tip of the tongue or top of the to-do list in the hospital setting. …
 By Kevin Rezzadeh
By Kevin Rezzadeh
Peer Reviewed
Injuries associated with amateur boxing include facial lacerations, hand injuries, and bruised ribs.1 While many of the superficial wounds and bone fractures can completely …
 By Brit Trogen
By Brit Trogen
Peer Reviewed
In 2001, the Institute of Medicine’s Crossing the Quality Chasm became the seminal paper recognizing patient-centered care as a crucial component of overall health care quality.[1] …
 By Dixon Yang, MD
By Dixon Yang, MD
Peer ReviewedÂ
AbstractÂ
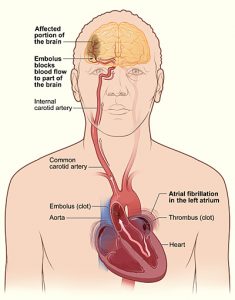
Atrial fibrillation (AF) is a common arrhythmia, especially in the elderly, and is often asymptomatic. However, absence of symptoms does not confer better prognosis. Many patients with AF …
 By Monil Shah, MD and Arun Manmadhan, MD
By Monil Shah, MD and Arun Manmadhan, MD
Peer Reviewed
A 64-year old male with a history of hypertension, dyslipidemia, and uncontrolled diabetes is brought to the emergency room with new …