 By Olivia Descorbeth
By Olivia Descorbeth
Peer Reviewed
As individuals advance in age, they tend to accumulate medical conditions that require a bevy of pharmaceutical treatments to manage. As a result, polypharmacy, generally defined as the use …

By Olivia Descorbeth
Peer Reviewed
As individuals advance in age, they tend to accumulate medical conditions that require a bevy of pharmaceutical treatments to manage. As a result, polypharmacy, generally defined as the use …
 By Marie T. Mazzeo
By Marie T. Mazzeo
Peer Reviewed
Anemia poses a significant threat to public health and affects approximately 2 billion people, nearly one-quarter of the world’s population.1,2 Iron deficiency is the most common cause of anemia worldwide and is the most common nutrient deficiency. …
 By Galen Hu
By Galen Hu
Peer Reviewed
It seemed like everyone I spoke to about heart attacks during my clinical year of medical school had a different opinion on the famous mnemonic “MONA BASHâ€:
Morphine
Oxygen
Nitrates
Aspirin/Anti-platelet
Beta-blocker
Angiotensin-converting enzyme …
 By Jonah Klapholz                             …
By Jonah Klapholz                             …
 By Mercedes Fissore-O’Leary
By Mercedes Fissore-O’Leary
Peer Reviewed
I.
It is his youngest’s birthday today. His oldest is in the military, like he was. Except he served in the navy, in Vietnam. He …
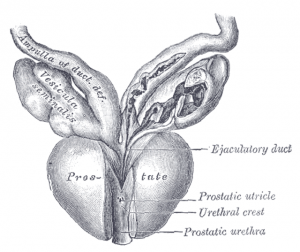 By Ian Jaffe
By Ian Jaffe
Peer Reviewed
Recent headlines about increasing rates of metastatic prostate cancer have had many patients asking if they should be tested.1 This article will review the history and role of …
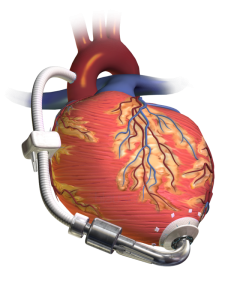 By Joshua Novack, MD
By Joshua Novack, MD
Peer Reviewed
Case: 74 year old male with a history of heart failure with reduced ejection fraction (EF 20%) diagnosed 10 years ago comes in with subacute progressive lower …
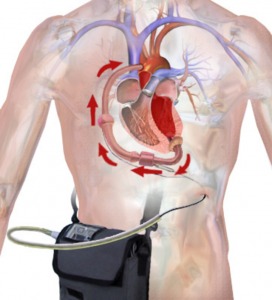 By Raymond Barry
By Raymond Barry
Peer Reviewed
A 2020 report published by the American Heart Association (AHA) in conjunction with the National Institutes of Health (NIH) found that an estimated 6.2 million American adults had …