 By Carolyn Akers
By Carolyn Akers
Peer Reviewed
Roger Daltrey, co-founder and lead singer of The Who, had this to say of bandmate Pete Townshend’s famous penchant for bludgeoning Fender Stratocasters:
“Pete wasn’t just smashing his guitar. He …

By Carolyn Akers
Peer Reviewed
Roger Daltrey, co-founder and lead singer of The Who, had this to say of bandmate Pete Townshend’s famous penchant for bludgeoning Fender Stratocasters:
“Pete wasn’t just smashing his guitar. He …
 By Olivia Descorbeth
By Olivia Descorbeth
Peer Reviewed
As individuals advance in age, they tend to accumulate medical conditions that require a bevy of pharmaceutical treatments to manage. As a result, polypharmacy, generally defined as the use of …
 By Matthew Auda
By Matthew Auda
Peer ReviewedÂ
Let’s begin with a case. The patient is a 67-year-old female with a past medical history of hypothyroidism referred to your Cardiology Clinic by her primary care physician …
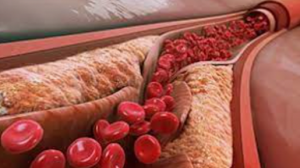 By Zachary Henig
By Zachary Henig
Peer Reviewed
Atherosclerosis is the primary risk factor for cardiovascular disease, the leading cause of mortality worldwide. To understand the pathophysiology of atherosclerosis, we turn to advances made in molecular …
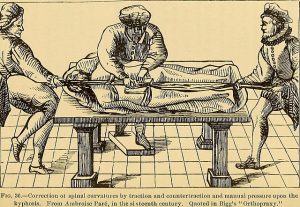 By: Michael Moore
By: Michael Moore
Peer Reviewed
“Too many complex back surgeries are being performed and patients are suffering as a result†wrote National Public Radio health science journalist Joanne Silberner in her 2010 article …
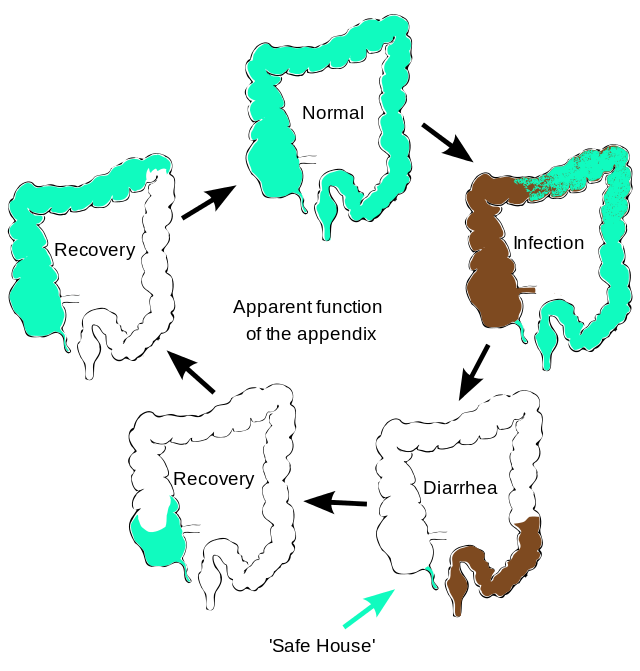 By Hannah Kopinsky, MD
By Hannah Kopinsky, MD
Peer Reviewed
Appendicitis is the most common reason for urgent surgery related to abdominal pain in the US, with a lifetime incidence of 8.6% for men and 6.7% for women.1 …
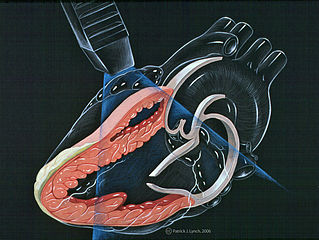 By Pamela Boodram, MD
By Pamela Boodram, MD
Peer Reviewed
A 68-year-old woman with a history of hypertension and well controlled type 2 diabetes presents to the ED with five days of progressively worsening dyspnea on exertion, …
 By Avani Kolla
By Avani Kolla
Peer Reviewed
During my trip to India, the “family bonding†reached a new level when I shared my upper respiratory infection with my parents and sister. On day two of …