 By Maria Garcia-Jimenez, MD/MHS, Abinav Baweja, MD, and Nicole LaNatra, MD
By Maria Garcia-Jimenez, MD/MHS, Abinav Baweja, MD, and Nicole LaNatra, MD
Peer Reviewed
Clinical vignette
A 65-year old woman with history of invasive breast cancer presents to her primary care provider for regular …

By Maria Garcia-Jimenez, MD/MHS, Abinav Baweja, MD, and Nicole LaNatra, MD
Peer Reviewed
Clinical vignette
A 65-year old woman with history of invasive breast cancer presents to her primary care provider for regular …
![]() Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Listen to 5Â Pearls segment of Iron Deficiency Anemia! By Dr. Cary Blum MD, Marty Fried MD and Shreya P. Trivedi MD; Illustration by Mike Natter MD
Time Stamps:

By Sara Stream, MD
Peer Reviewed
As resident physicians, we are taught to supplement serum potassium to a goal level of 4.0 mEq/L in all hospitalized patients. While the dangers of severe potassium abnormalities are …
![]()
Listen to CORE IM’s first 5 Pearls segment on Headaches!
Time Stamps
 By Helen Ma, MD
By Helen Ma, MD
Peer Reviewed
Our new Spotlight series uses case vignettes to explore diagnosis, pathophysiology, and management of a wide variety of diseases seen in the outpatient and inpatient settings. …
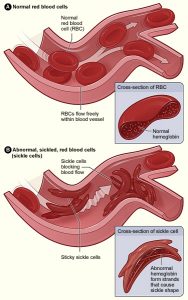 By Leonard Naymagon, MD
By Leonard Naymagon, MD
Peer Reviewed
Vaso-occlusive crisis (VOC), or pain crisis, is the most common clinical manifestation of sickle cell disease (SCD) and is responsible for the majority of emergency department (ED) visits …
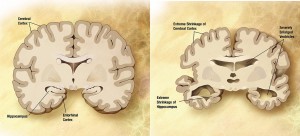 By Karen McCloskey, MD
By Karen McCloskey, MD
Peer Reviewed
In May 2012, The U.S. Department of Health and Human Services unveiled the “National Plan to Address Alzheimer’s Disease,†in response to legislation signed by President …
 By Eric Jeffrey Nisenbaum, MD
By Eric Jeffrey Nisenbaum, MD
Peer Reviewed
Mr. O is a 93-year-old man with a past medical history notable for severe Alzheimer’s dementia and amputation of the left upper extremity secondary …